Alternatives to UGENE
Draft
to do
- update links
For instructional purposes, I recommend use of UGENE as your genomics workbench for the semester. The software runs on macos, Win11, and Linux.
We use UGENE to
- apply and view sequence alignments
- Clustal Omega
- MUSCLE
- T-Coffee
- build gene (phylogenetic) trees
- IQTREE
- NJ
- PhyML
- mrBayes
- and to conduct basic descriptives for comparisons among our Project essential thirteen + 1 species
UGENE is a great graphical user interface (GUI) and comes with most of the bioinformatics software we need, ready to go without further installation on our part. However, some of you are new to installing software, and, particularly on macs, find UGENE confusing to install and run. My advice, please stick with it, let me know if you have difficulty with the software.
Alternatives to UGENE
Run all analysis in your browser, no software installation
But, if you’ve given up, or more importantly, you do not have a suitable computing platform (iPad or other tablet, Chromebook), then there are alternatives. The first option — NG Phylogeny website — is recent, and works very nicely — so well in fact you may prefer it to UGENE! The second option is a do it yourself option, where your work flow takes you to different web sites to accomplish what you need.
Both options require you to use Batch Entrez to download a FASTA file with your 13+1 protein sequences — see Download sequences with Batch ENTREZ . These alternatives are virtually foolproof, but I have found their use less attractive for my students — students have to bounce from one website to another, downloading and uploading files as they go — but as of Spring 2025, these procedures work.
NG Phylogeny website option
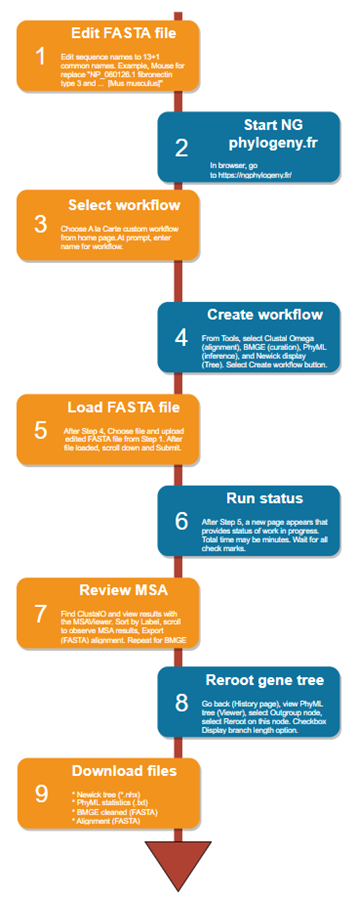
| 1. Download and save a txt file with all accession files in FASTA format — see Batch Entrez for help. See
2. Edit FASTA file. Edit sequence names to 13+1 common names. Example, Mouse for replace “NP_080126.1 fibronectin type 3 and … [Mus musculus]” 3. Select workflow. Choose A la Carte custom workflow from home page. At prompt, enter name for workflow. 4. Create workflow. From Tools, select Clustal Omega (alignment), BMGE (curation), PhyML (inference), and Newick display (Tree). Select Create workflow button. 5. Load FASTA file. After Step 4, Choose file and upload edited FASTA file from Step 1. After file loaded, scroll down and Submit. 6. Run status. After Step 5, a new page appears that provides status of work in progress. Total time may be minutes. Wait for all check marks. 7. Review MSA. Find ClustalO and view results with the MSAViewer. Sort by Label, scroll to observe MSA results, Export (FASTA) alignment. Repeat for BMGE. 8. Reroot gene tree. Go back (History page), view PhyML tree (Viewer), select Outgroup node, select Reroot on this node. Checkbox Display branch length option. 9. Download files
|
 |
Do it yourself options
1. The most general option is to rely strictly on online websites.
- Align sequences with Clustal Omega – link to server
- tree building
- PhyML tree building, link to server
- mBayes tree
- NJ tree building, link to server
- IQTREE, link to server
- Tree viewers
- icytree.org, view but no option to re-root trees, link to server
- iTOL interactive tree of life, view and re-root tree files, link to server
- TreeDyn, view and re-root tree files, link to server
Certainly an option for students who do not wish to (or cannot) download and manage software, and our primary option for ChromeBook users (Jalview and R the other options), but website option requires user to learn multiple interfaces, requires registration (for some sites), interfaces change, browser support may be an issue, and is subject to internet connectivity issues.
Like the detailed example I provided for UGENE (Try an example to learn UGENE), I’ve added a corresponding example to run same analysis on browser-only apps (Alternative to UGENE – Try an example to learn).
2. Download and install OTHER software instead of UGENE. Note: these options require you to install software to your computer, so would not be options for Chromebook.
- Seaview Multiplatform GUI for molecular phylogeny (click here for DrD’s seaview page). Although the interface is not modern looking, this is your best alternative if you cannot get UGENE to work for you
- MEGAX – link to homepage.This option has many good things about it, particularly how it handles tests of molecular clock model, but it is primarily designed for Windows PCs and there’s now a good macos version. However, the workflow used by MEGAX is confusing, with many different windows and files that the user has to keep track of.
- NCBI Genome Workbench – link to homepage. You’d think this would be the best option. However, compared to UGENE, the performance of this app is very poor. My guess is this has to do with networking issues, as I have better luck from home than on campus with UGENE. While gbench is great for viewing and analyzing nucleic acids, much more limited for work with proteins. Limited alignment algorithms, limited tree-building algorithms.
- Jalview. Just now came across this package. Looks to be a viable alternative. More to follow. jalview.org
- R and packages (ape, ggmsa), and Bioconductor packages (msa). This is a good the option, but the installation can be challenging, recommended for users more comfortable with their computers. However, the payoff is that the user ends up with an analytical platform more powerful than UGENE.
R would be the best choice, and I do provide R instructions on many of the pages. See R installation page and links here.
/MD