RefSeq, NCBI‘s Reference Sequence project, is a non-redundant, annotated and curated set of sequences that serve as reference standards.
Model RefSeq: RNA and protein products that are generated by the eukaryotic genome annotation pipeline. These records use accession prefixes XM_, XR_, and XP_.
Known RefSeq: RNA and protein products that are mainly derived from GenBank cDNA and EST data and are supported by the RefSeq eukaryotic curation group. These records use accession prefixes NM_, NR_, and NP_ .
Bioinformatics I: BLAST
As always, evidence is expected in your Lab Notebook
Objectives:
- Demonstrate how to retrieve results from a BLAST search.
- Describe elements of sequence as FASTA format.
- Define key bioinformatic terms.
- Demonstrate use of text only data sets.
- Examine and interpret results from BLAST search.
- Demonstrate and interpret changes to software settings.
- Collect proper evidence from work, documented in Notebook.
- Use Blast Entrez to retrieve sequences.
Overview:
This Worksheet has seven (7) self-study questions.
Prepare your responses to the questions in your Digital NoteBook. Follow all general instructions about how to provide lab notebook evidence. For example, show your work in your notebook, include intermediate steps (e.g., screenshots of settings, results).
Submit completed worksheet:
You will need to create a docx or pdf file with your work. This is easy to do in OneNote. Once your edits are complete,
- select to print the notebook page(s) (Windows: Ctrl+P, macOS: ⌘+P)
- rename the file appropriately (name course task)
- submit your pdf file to this Canvas page.
BLAST work
Billions of protein and nucleic acid (DNA and RNA) sequences are stored in publicly accessible databases. For researchers with an unknown sequence, the first step is to query these databases to see if their unknown sequence “matches” known sequences.
The Basic Local Alignment Search Tool (BLAST) is a program used to find regions of local similarity between protein or nucleotide sequences. BLAST compares nucleotide or protein sequences to sequences stored in a database; BLAST then calculates statistical significance of the matches.
For background, here’s a link to BLAST glossary http://www.ncbi.nlm.nih.gov/books/NBK62051/ . Don’t forget Wikipedia!
Question 1. Provide a definition IN YOUR OWN WORDS for each of the following terms.
- accession
- refseq
Question 2. What is meant by the term “Local alignment”?
BLAST
To access BLAST go to http://blast.ncbi.nlm.nih.gov/ (or simply search “BLAST” on Google). Figure 1, a screenshot of BLAST Home page, Spring 2022. Not shown, Standalone and Specialized search BLAST icons.
Figure 1. Screenshot, upper portion of NCBI BLAST homepage (Spring 2022)
BLAST front-page options
As you can see from the front page, BLAST now has many options. If the goal is to compare sequences from one of the available published genomes, then you would select BLAST Assembled RefSeq Genomes. Of particular note is the addition of many specialized BLAST tools, including Primer-BLAST which would be used to help you design or check PCR primers.
However, in general, you will be selecting from the Basic BLAST programs. For example, if working with DNA, then select blastn (nucleotide blast). If working with protein sequences, then use blastp (protein blast).
Regardless of the BLAST selected, the basics are similar enough. Let’s try a worked example, mystery sequence 1.
First, the BLAST program expects the sequence in a certain format (FASTA). Here’s one for you
>mystery sequence 1 TAATGACCCGCTGGTCCTGAGGAAGAGGTGCTGACGACCAAGGAGATCTTCCCACAGACCC AGCACCAGGGAAATGGTCCGGAAATTGCAGCCTCAGCCCCCAGCCATCTGCCGACCCCCCC ACCCCAGGCCCTAATGGGCCAGGCGGCAGGGGTTGAGAGGTAGGGGAGATGGGCTCTGAG ACTATAAAGCCAGCGGGGGCCCAGCAGCCCTCAGCCCTCCAGGACAGGCTGCATCAGAAGA GGCCATCAAGCAGGTCTGTTCCAAGGGCCTTTGCGTCAGGTGGGCTCAGGATTCCAGGGTG GCTGGACCCCAGGCCCCAGCTCTGCAGCAGGGAGGACGTGGCTGGGCTCGTGAAGCATGT GGGGGTGAGCCCAGGGGCCCCAAGGCAGGGCACCTGGCCTTCAGCCTGCCTCAGCCCTGC CTGTCTCCCAGATCACTGTCCTTCTGCCATGGCCCTGTGGATGCGCCTCCTGCCCCTGCTGG CGCTGCTGGCCCTCTGGGGACCTGACCCAGCCGCAGCCTTTGTGAACCAACACCTGTGCGG CTCACACCTGGTG GAAGCTCTCTACCTAGTGTGCGGGGAACGAGGCTTCTTCTACACACCC AAGACCCGCCGGGAGGCAGAGGACCTGCAGGGTGAGCCAACTGCCCATTGCTGCCCCTGG CCGCCCCCAGCCACCCCCTGCTCCTGGCGCTC
Note: I’ve confirmed the mystery sequence runs correctly in BLAST. However, if you copy and paste this sequence into “Enter Query Sequence” BLAST window, run BLAST, then get an error message like: “Message ID#33 Error: Query contains no data: Query contains no sequence data,” this is due either to a missing EOL (end of line separators, hard return) for the first line, or EOL or SPACES introduced at the end of the lines containing the sequence. BLAST expects a single comment line, with the next and all subsequent lines consisting of one uninterrupted sequence. If BLAST does not run having used COPY/PASTE within your browser, suspect one or both errors. Use a text editor to check for hidden “non-printable” characters (spaces, EOL, etc) to fix your FASTA file. Use of text editor to find and replace non-printable characters was presented in Text files are data files.
Question 3. Does this “mystery sequence” call for use of blastn or blastp?
<h4align=”left”>
You can see that FASTA format allows a comment line marked by “>” at the start, then beginning with the second line follows with the sequence data. You can copy and past this sequence into the BLAST window
Like most searches, start with the Default” options — in other words, paste the sequence into BLAST, Press the “BLAST” button to start the search (Fig. 2). Here’s what we get for this sequence
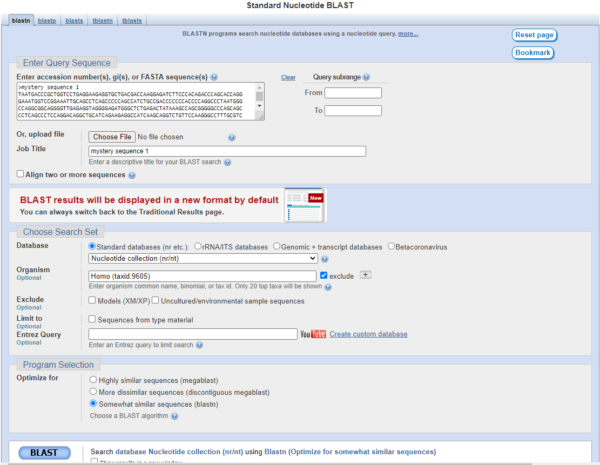
Figure 2. Screenshot of our mystery sequence basic search, humans selected as target organism.
If you run into trouble (i.e., the BLAST returns no results, then you go back and modify parameters to the algorithm (i.e., click on the link +Algorithm parameters).
While BLAST is running, this screen updates in your browser window (Fig. 3).
Figure 3. Screenshot BLAST running status page
When BLAST is done you get this page (Fig. 4, partial screenshot, the actual page continues down the screen). The default returns up to 100 possible matches. I’d like you to record the first and the last match.
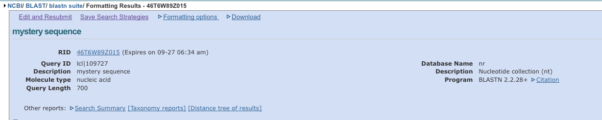
Figure 4. Partial screenshot BLAST results page, top portion; the actual page continues down the screen
Scroll down or click on Description tab (Fig. 5)
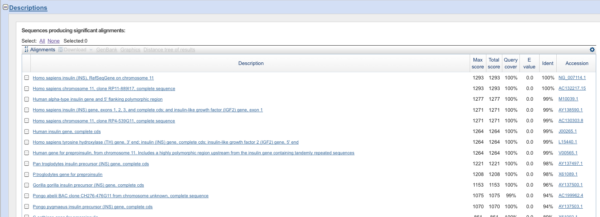
Figure 5. Screenshot portion of BLAST results page, description
Click on Graphic Summary tab to view (Fig. 6).
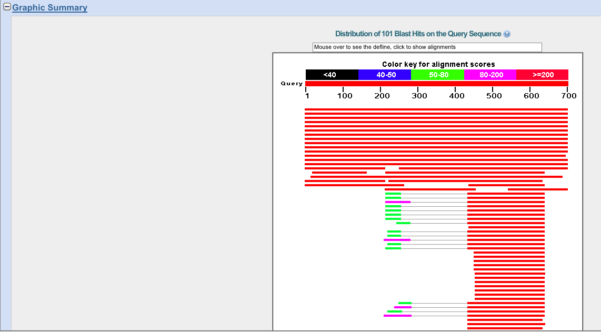
Figure 6. Screenshot of graphical summary tab, results page BLAST
Each of the screen panels provides information, but the DESCRIPTION page is the one we want — we want to know, is there a sequence that matches our mystery sequence?
In Figure 7, I enlarged the first line from Description tab shown in Figure 5. (Remember, the output here is just an example, it is not your example.)
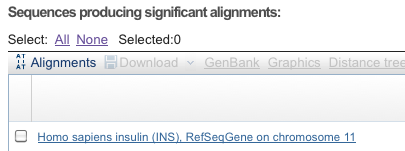
Figure 7. Screenshot, portion of Description tab displayed in Figure 5.
We see that BLAST has returned its first, and therefore, most likely hit, human Insulin. Continuing off the the right of this line shown in Figure 5, the supporting evidence for the match is visible (Fig. 8).
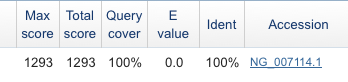
Figure 8. Screenshot supporting evidence for the match, visible at right-hand portion of image displayed in Figure 7.
The E-value (Expected value) is equivalent (though not exactly so) to the p-value from a statistical test. Like statistical tests, we interpret low E-values that the chance of our sequence matching Insulin by chance alone is highly unlikely. In our example it reported an E-value of “0.0” which is misleading — as you know, an empirical probability cannot be exactly zero (or for that matter, exactly one — there’s always chance deviations). BLAST is reporting “0.0” because it is rounding the value — actually the E-value is really really small, but not zero.
To help us interpret our results, BLAST also provides the inverse of the E-value or “Identity” and we see that our mystery sequence matches by 100% over the range of sequence searched by BLAST.
Question 4 What is the percent identity for our mystery sequence with Pan troglodytes gene for insulin precursor?
Question 4a) What is a Pan troglodytes?
Question 5. Return to the BLAST search page (back arrow in browser will do) then select +Algorithm parameters. Locate the “Word size” value (report it), then change it from the default to
a) a much larger value
b) a much smaller value
then for (a) and (b) re-run the BLAST search for mystery sequence 1 and report on the first match, if any, and the last match, if any– do you get the same answer as before?
In question 5 I had you explore effects of changing the “word size” on the search. The “word size” refers to how many letters the algorithm starts the search. For example, if word size was “3”, then BLAST would search a sequence of TTGGCCGAGAGACCT in blocks of threes TTG GCC GAG AGA CCT. It would find matches in the database for each of these three letter combinations (a lot of sequences!), then next iteration start to search against larger word combinations.
Question 6. What effect does word size have on search results?
What else can you do from the BLAST results screen?
Our goal was to check for a match between our mystery sequence and a known sequence in a database. We received matches with 100% identity and conclude that our mystery sequence was human insulin. So, we’re done. However, a number of things could follow. A useful visual display can be had by first selecting some of the sequences (click in the little box next to each line you are interested in), then choosing “Distance tree of results”. In addition to our human insulin sequence I selected several other primate sequences to generate the following distance tree (Fig. 9).
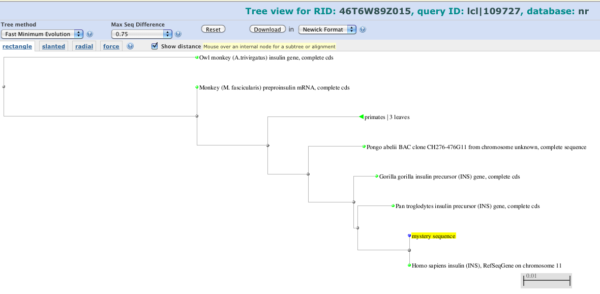
Figure 9. Screenshot of distance tree made from BLAST aligned sequences.
Now, distance has a precise definition, but for now, think of it like this. If two things are identical, then the distance between them is small (zero if truly identical). If two things are different, then the distance between them is large, proportional to how different they are. In the image our “mystery sequence” is noted by yellow tag and it shares a branch with the human insulin gene. The branch length is short, so this is in keeping with our 100% sequence match.
Question 7. Try this mystery sequence yourself.
>mystery sequence 2
MEGAGGANDKKKISSERRKEKSRDAARSRRSKESEVFYELAHQLPLPHNVSSHLDKASVMRLTISYLRVRKLLDAV
(a) which blast search algorithm should you use blastn or blastp?
(b) what is the accession number of the top match?
(c) what species and what gene is the top match for?
(d) generate a distance tree for the top ten matches.
(e) based on your experience/results from Question 5, select a new word size and re-run the BLAST search on mystery sequence 2 and repeat question 7b-d.
Use Blast Entrez to retrieve sequences
Once you’ve built up a list of accession numbers, you can retrieve the sequences via Batch Entrez. See Download sequences with BATCH Entrez for help.
/MD