Make a phylogeny from morphological characters
This page and guide is intended for beginning students, therefore the cases described do not address all issues related to the problem.
From character matrix to phylogeny
This page contains two approaches to build a phylogenetic tree.
One approach uses the statistical programming language R and specialized R packages. I provide a working script on this page — small modifications only needed to reflect your data — the script should run in a standard R installation on your computer or in the cloud, eg, Google CoLab.
The second approach, listed first, uses a simple but powerful set of applications at a web server in Canada: https://trex.uqam.ca/.
Select tree inference from the Main Menu (Fig 1).
Figure 1. Screenshot trex home page.
Step 1. Before running the software you must have your data in the correct format.
Convert character matrix to phylip format.
Recall, a character matrix looks like
taxa, trait01, trait02, trait03, trait04, trait05, trait06 spp01, 0, 0, 0, 0, 0, 0 spp02, 1, 1, 1, 0, 0, 0 spp03, 0, 1, 1, 0, 0, 1 spp04, 1, 0, 0, 0, 0, 0 spp05, 1, 1, 1 ,1, 0, 0 spp06, 1, 1, 1, 1, 0, 1
phylip file format looks like
6 6 spp01 000000 spp02 111000 spp03 011001 spp04 100000 spp05 111100 spp06 111101
About the format. Line one begins with the number of taxa (6 in this example) followed by a space then the number of characters (again, 6 in this case). The next line begins with the taxa name (no spaces or special characters) and cannot be longer than ten characters. Strict phylip format expects character states to start in column 11 (as this example does). To help you view this I show entry for spp2 by column number.
1234567891011 spp02 111000
For few characters and taxa, it’s pretty easy to convert by hand. For larger files, see Dohm’s R script at ccc.
From trex, select tree inference, NJ for Neighbor Joining, and other, eg., PARS(PHYLIP) (Fig 1).
Copy/paste phylip file into the text box, then select the Compute button (Fig 2).
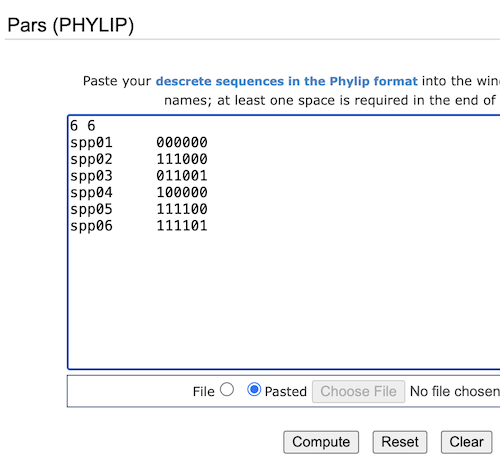
Figure 2. Screenshot of trex-online PARS window with phylip-stype character matrix.
If all goes well, no error with the input file, then the output screen appears (Fig 3). If the page returns an error message, then the problem is most likely with the phylip format.
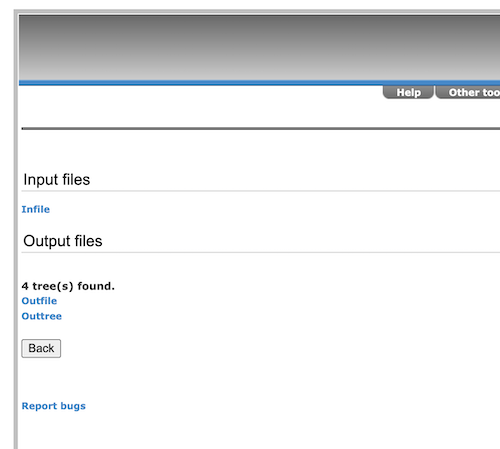
Figure 3. Screenshot trex-online results window.
Note that four trees were found, not one single tree. When a single tree is produced, tree viewer will automatically display the one tree. Here, with the parsimony option, four equally likely trees were produced. We need to do a little more work to view one of the trees.
Click on the Back button and navigate to Tree viewer. Alternatively, click on Outtree link (Fig 3, blue under 4 tree(s) found). The following four lines of Newick code are listed (Fig 4).
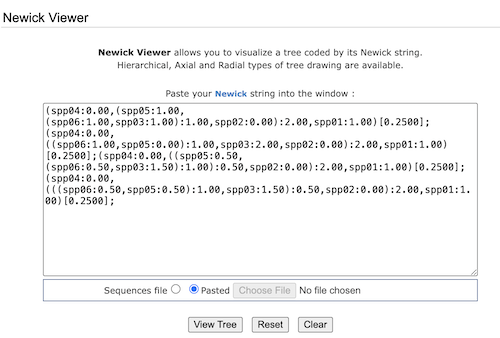
Figure 4. Screenshot trex-online Tree Viewer window with multiple lines of Newick code. each line represents a tree.
Copy all four to your notebook, then select one to view, deleting all the rest from the Tree Viewer window (Fig 5). The code is not ready — if you click View Tree, an error is generated. You need to edit the code.
![Screenshot trex-online Tree Viewer window with only the first tree. Note that the code needs editing - remove the [0.2500] at the end, but retain the ending semi colon ;.](https://geneticslab.letgen.org/wp-content/uploads/2025/09/trex_tree_viewer_oneFile_noEdits.png)
Figure 5. Screenshot trex-online Tree Viewer window with only the first tree. Note that the code needs editing – remove the [0.2500] at the end, but retain the ending semi colon “;”.
The error is because of additional code added near the end of the Newick file. All Newick code end with a semicolon, it’s the [0.2500] that causes the error. Your edited Newick code for this tree should read
(spp04:0.00,(spp05:1.00,(spp06:1.00,spp03:1.00):1.00,spp02:0.00):2.00,spp01:1.00);
With this edit completed, View Tree returns Figure 6.
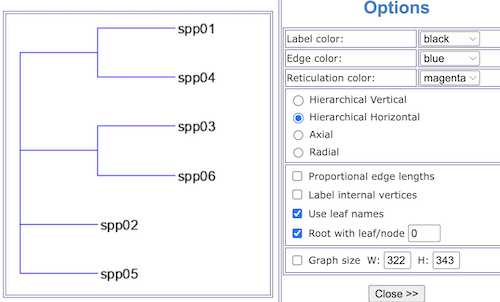
Figure 6. Screenshot, the parsimony tree.
Repeat steps above for other tree algorithms, for example, Neighbor Joining.
Click here to review R script for neighbor joining or parsimony analysis based on morphology character matrix.
/MD