Working with Newick file format
to do
- update links
images- Update to current UGENE version
There are several formats for representing tree diagrams, but Newick and Nexus are among the simplest and most widely used phylogeny formats. The basic grammar of these formats is that parentheses are used to group taxa, commas are used to branch taxa, and colon followed by number sets the branch length. But for now, you’re here because you are trying to view a tree file in UGENE or another tree viewer app.
Save a tree file without using UGENE
Most of the tree files you will work with in this course you generate within UGENE. However, in some cases, e.g., Our consensus phylogenetic tree, you are given the code for the tree and you simply need to grab the code from the webpage and input it into UGENE or some other tree viewer app.
If the code is on a webpage, the simplest thing to do is to
- Highlight and copy the Newick code to your clipboard
- start your text editor (Text files are data files)
- paste the code into the your text editor
- save the file as text only (default for NotePad, TextEdit default is rich text or .rtf, not text only — see Text files are data files for instructions)
- I recommend saving the file with .nwk extension, not .txt, your text editor default.
Now that you have the file, you can then view the tree in one of many available tree viewers, e.g., IcyTree, or an even more powerful viewer at iTol: Interactive Tree Of Life, or even with UGENE. To import the file into UGENE, go to File > Open as… , then select your new file (eg, example “consensus.nwk”) (Fig 1).
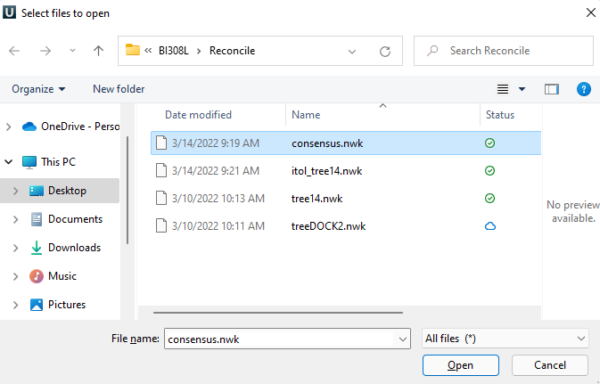
Figure 1. Screenshot of UGENE Open as… File Explorer dialog on Win11 PC.
Next, UGENE will ask you to identify the file type (Fig 2). Note that UGENE should recognize the format, so you should only have to confirm the choice.
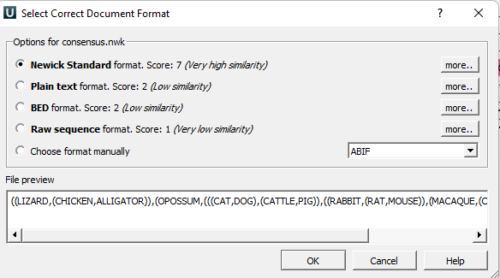
Figure 2. Screenshot of second menu request to identify file type to complete UGENE Open as… Note there are no branch lengths included in this Newick code example.
Click OK button, and the file will be added to UGENE Project, which you can confirm in the Project Window (Fig 3),
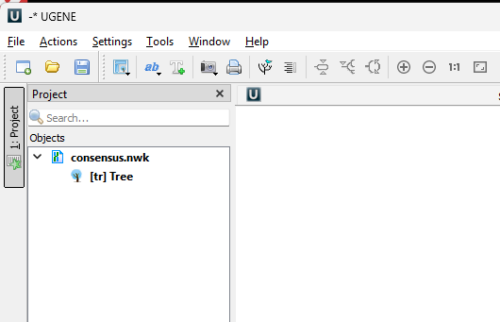
Figure 3. Screenshot UGENE project window.
and the tree window should open to display your new tree file (Fig 4).
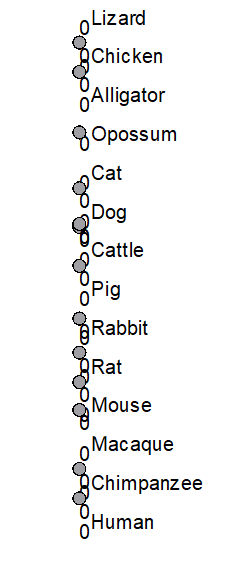
Figure 4. UGENE Tree View of our imported Newick file (See Figure 3 for hint)
Wait! The tree I imported into UGENE looks strange!?
UGENE can work with Newick and Nexus files, but UGENE unfortunately can’t handle some of the variants of the formats. For example, our imported tree (Fig 1-3), shown in Figure 4, lacks that distinctive branching pattern we have come to expect. We haven’t done anything wrong, this is a limitation of the current version (41) of UGENE. UGENE doesn’t know how to handle tree files which lack branch lengths (note the “0” in Fig 4). If you add branch lengths, UGENE will display the tree file correctly (Fig 5).
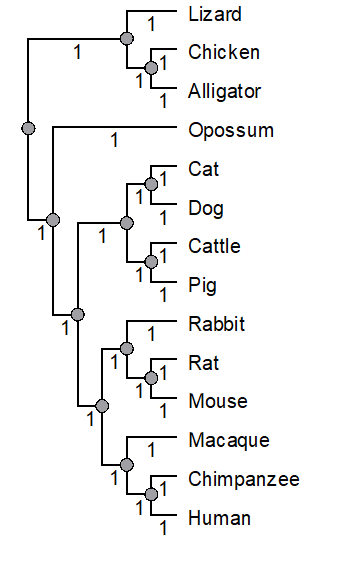
Figure 5. After setting the branch lengths equal to one, UGENE Tree View correctly displays our imported Newick file
Remember, Newick files are just text files, so you can add the branch lengths in by hand (paying attention to Newick grammar, see below), or better, use another app like FigTree. I used FigTree and the option to Transform the branch lengths to equal length.
For another example, the NCBI TreeViewer may include names for internal nodes, like Carnivora, which seems to cause problems for UGENE. Here’s what a Newick file looks like from NCBI TreeViewer.
(Anolis_carolinensis:4, ((Rattus:4, Mus_musculus:4)Muridae:4, Oryctolagus_cuniculus:4, Bos_taurus:4, Sus_scrofa:4, (Felis_catus:4, Canis_lupus_familiaris:4)Carnivora:4, ((Homo_sapiens:4, Pan_troglodytes:4)Hominidae:4, Macaca:4)Primates:4, Didelphis_virginiana:4)Mammalia:4, Gallus_gallus:4, Alligator:4)Chordata:4;
This is an acceptable Newick file. Copy and paste it into a text file, call it tree.nwk.
You can check your Newick files rapidly at any number of online sites, for example https://icytree.org/, or an even more powerful viewer at iTol: Interactive Tree Of Life. If you copy and paste the above information into icytree (File → Enter tree directly…), you’ll find it renders perfectly.
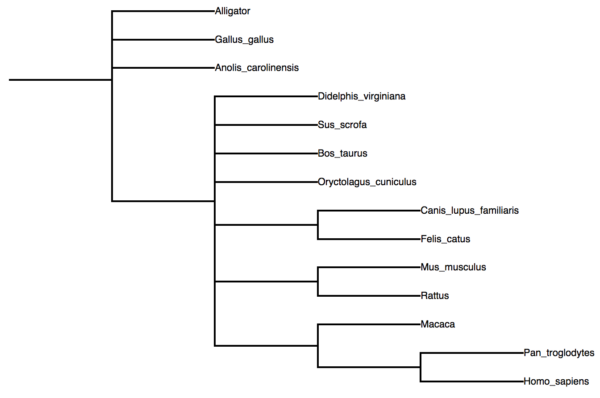
Figure 6. A nice tree from our Newick file, courtesy of icyTree.org
Similarly, if you choose iTOL, select Upload from the menu, copy Newick code from your file (i.e., have it open in your text editor a tree
It is simple enough to edit the newick file to suit UGENE. Open your Newick file in your favorite text editor (again, it’s just a text file!), and perform a little surgery. While a bit of a pain, it is straight-forward. Note: If you edited your sequence names in UGENE, e.g., changed Anolis_carolinensis_(green_anole) → Lizard, then you don’t need to edit your Newick file in a text editor
Edit your Newick file from NCBI TreeViewer
Step 1. Open your .nwk file into your favorite text editor. A screenshot of tree.nwk in TextEdit is shown in Figure 7.
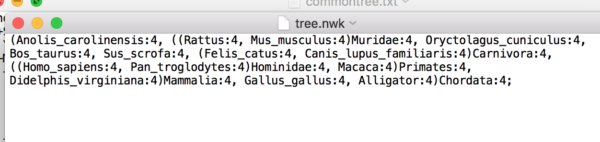
Figure 7. Screenshot of tree.nwk in macOS TextEdit.
Step 2. Look for lower taxonomic rank names immediately following a “)” . For example, scan the first line from left to right and find “…4)Muridae”. Delete “Muridae”. Continue and remove “Carnivora,” etc. I highlighted these names in red for you (Figure 8).
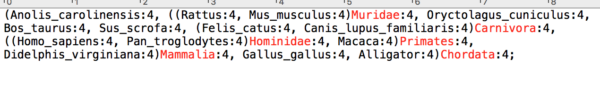
Figure 8. Names to remove shown in red.
Note that if you checked the lower rank option at NCBI Taxonomy Browser, then you may have additional names to remove. After
Step 3. After deleting those names, your file should look like the one in Fig. 9. You can now load this file into UGENE with no further difficulty.
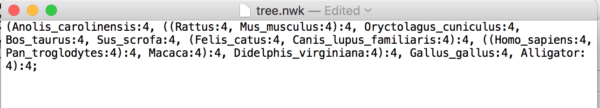
Figure 9. Newick file with edits, suitable for viewing in UGENE.
Step 4. You can also change from the scientific names to common names, simply edit the Newick file to make the changes (Fig. 10).
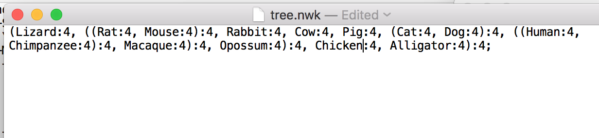
Figure 10. Our Newick file after removing incompatible lower rank names and changing species names to common names.
That’s it. UGENE will now be happy with this Newick-format.
Questions
Quiz: Trees and Newick files
References
Czech, L., Huerta-Cepas, J., & Stamatakis, A. (2017). A critical review on the use of support values in tree viewers and bioinformatics toolkits. Molecular biology and evolution, 34(6), 1535-1542. https://doi.org/10.1093/molbev/msx055
Felsenstein, J. (2014). PHYLIP (phylogeny inference package), version 3.698. Joseph Felsenstein. https://evolution.genetics.washington.edu/phylip.html
Guindon S., Gascuel O. 2003. A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology 52:696–704. https://doi.org/10.1080/10635150390235520
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., & Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic biology, 59(3), 307-321. https://doi.org/10.1093/sysbio/syq010
1990, Gary Olsen’s Interpretation of the “Newick’s 8:45” Tree Format Standard Available from: http://evolution.genetics.washington.edu/phylip/newick_doc.html, retrieved 11 March 2019
Huelsenbeck, J. P., & Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics, 17(8), 754-755. https://doi.org/10.1093/bioinformatics/17.8.754
Letunic, I., & Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics, 23(1), 127-128. https://doi.org/10.1093/nar/gkab301
Maddison, D. R., Swofford, D. L., & Maddison, W. P. (1997). NEXUS: an extensible file format for systematic information. Systematic biology, 46(4), 590-621. https://doi.org/10.1093/sysbio/46.4.590
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., & Lanfear, R. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular biology and evolution, 37(5), 1530-1534. https://doi.org/10.1093/molbev/msaa015
Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics, 13(5), 555-556. https://doi.org/10.1093/bioinformatics/13.5.555